Metformin Isnt What They Told You MOTS-C Is The Real Deal
MOTS-C vs. Metformin: Why This Mitochondria-Derived Peptide Is Rewriting Longevity Science
For years, Metformin has been hailed as the gold standard anti-aging compound — prescribed by longevity clinics, celebrated by biohackers, and quietly added to the daily regimens of some of the wealthiest people on earth. But emerging research suggests this narrative is built on incomplete science, and a far more targeted compound called MOTS-C may be addressing the actual root causes of aging that Metformin simply cannot touch. In this article, we break down the three core molecular mechanisms of aging, explain exactly why Metformin falls short on each of them, and explore the evidence supporting MOTS-C as a genuinely comprehensive longevity peptide.
What Aging Actually Is: The Three Core Molecular Mechanisms
Before evaluating any longevity compound — whether it is Metformin, MOTS-C, Rapamycin, or anything else — it is essential to understand what aging actually is at a molecular and cellular level. Aging is not a single disease or a single symptom. It is the accumulated, progressive deterioration across three distinct but deeply interconnected biological mechanisms. Address all three and you meaningfully extend both lifespan and healthspan. Miss even one and you are, at best, slowing the clock while the foundation continues to crumble.
Mechanism 1: Genomic Instability and DNA Damage Accumulation
Every single day, each of your cells sustains between 10,000 and 100,000 DNA lesions. These lesions originate from oxidative stress, background radiation, spontaneous chemical reactions, and errors during DNA replication. Your cells are equipped with sophisticated repair machinery — including nucleotide excision repair, base excision repair, mismatch repair, and homologous recombination — designed to catch and correct these errors before they propagate. They also deploy autophagy and mitophagy to remove irreparably damaged components.
However, these systems are not perfect. Over time, some damage escapes repair and accumulates in the genome. A landmark 2019 study published in Nature Reviews Cancer confirmed that organisms with lower spontaneous mutation rates live significantly longer. Humans outlive mice not primarily because of body size, but because our DNA repair mechanisms are more robust and our mitochondria generate far less reactive oxidative stress per unit of time.
When DNA damage accumulates unchecked, three catastrophic downstream events unfold. First, tumor suppressor genes like p53 — often called the "guardian of the genome" — become mutated, removing the cell's ability to detect damage and trigger apoptosis, which allows cancer to develop. Second, a genomic instability cascade occurs: one mutation can compromise a DNA repair gene, making the cell progressively worse at fixing subsequent damage and accelerating mutation rates exponentially. Third, damaged cells trigger cellular senescence — a cellular checkpoint that halts division to prevent further genomic error propagation.
The primary enemy driving genomic instability is reactive oxygen species (ROS) — specifically superoxide radicals produced when electrons leak from Complexes I and III of the mitochondrial electron transport chain. These superoxide radicals react directly with DNA, creating the very lesions that accumulate over a lifetime. Your mitochondria, in other words, are simultaneously your primary energy source and your primary source of genomic damage.
Mechanism 2: Mitochondrial Dysfunction and ATP Failure
Mitochondria are not simply cellular organelles — they are endosymbiotic bacteria that were integrated into eukaryotic cells billions of years ago. They retain their own circular DNA (approximately 16,500 base pairs in humans), their own ribosomes, and they replicate independently of the cell cycle. They produce ATP through oxidative phosphorylation: electrons flow sequentially through Complexes I, II, III, and IV of the electron transport chain, pumping protons across the inner mitochondrial membrane to build a proton gradient. Complex V (ATP synthase) then uses that gradient to phosphorylate ADP into ATP — the universal energy currency powering every cellular process in your body.
As aging progresses, mitochondria become progressively dysfunctional. Electron transport chain proteins accumulate oxidative damage. The lipids in the inner mitochondrial membrane become oxidized and lose their structural fluidity. Mitochondrial DNA accumulates mutations faster than repair systems can correct them. Mitochondrial ribosomes become less efficient at synthesizing the proteins needed to maintain the electron transport chain itself.
The outcome is measurable and devastating. A 2018 study published in Aging Cell measured mitochondrial ATP production across subjects aged 30 to 80 and found that every decade of aging produced approximately a 10% decline in maximum mitochondrial ATP output. By age 80, subjects possessed roughly 50% less mitochondrial ATP production capacity than they had at age 30. This is not an inevitable consequence of calendar age — it is the direct result of accumulated mitochondrial damage that was not adequately repaired, cleared, or replaced. It explains why aging individuals experience chronic fatigue, cognitive decline, muscle atrophy, and declining cardiovascular capacity. Their cells are literally running on insufficient fuel.
Mechanism 3: Cellular Senescence and the SASP Inflammatory Cascade
Every human cell has a finite replication capacity — approximately 50 to 70 divisions before it enters a state called senescence. This limit, known as the Hayflick Limit, exists as a tumor suppression mechanism. Once a cell has divided enough times that genomic errors have meaningfully accumulated, it must stop dividing to prevent cancerous proliferation. When this checkpoint is triggered, the cell enters senescence: it stops dividing, but it does not die.
Senescent cells — frequently called "zombie cells" in popular science — remain metabolically active within your tissues but serve no functional purpose. Worse, they produce what is known as the Senescence-Associated Secretory Phenotype (SASP), a toxic cocktail of inflammatory cytokines and proteases including TNF-alpha, IL-6, IL-1 beta, and matrix metalloproteinases (MMPs). These factors leak continuously into surrounding tissue, damaging healthy cells, promoting fibrosis, driving chronic inflammation, and degrading the extracellular matrix.
By age 70 to 80, approximately 30% of cells in certain tissues may be senescent, depending on tissue type. The accumulation of these cells is one of the strongest predictors of accelerated biological aging. A 2019 study in Nature Medicine demonstrated that clearing senescent cells in aged mice extended both lifespan and healthspan by 35% — improving cognition, cardiovascular function, and metabolic health simultaneously. Senolytic intervention — the targeted elimination of senescent cells — stands as one of the most validated anti-aging strategies in current research.
Why Metformin Falls Short as a Longevity Drug
Metformin is a biguanide compound with origins dating to the 1920s and formal approval for type 2 diabetes treatment in the 1950s. It remains the most widely prescribed diabetes medication globally, and in recent decades it has been aggressively repurposed as an anti-aging compound by longevity clinics and high-profile biohackers. But the scientific case for Metformin as a longevity drug is far weaker than its popular reputation suggests.
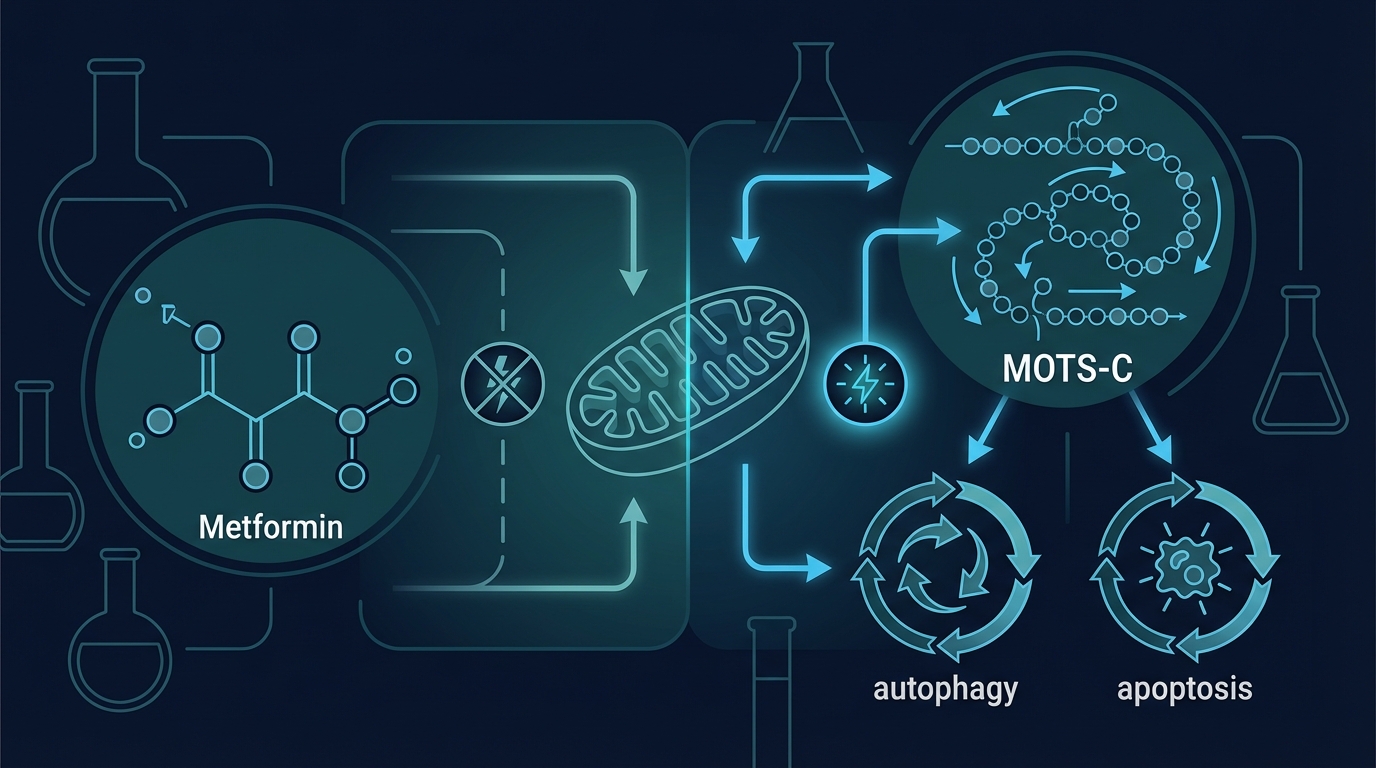
Metformin's primary mechanism of action involves the inhibition of Complex I of the mitochondrial electron transport chain. By partially blocking Complex I, Metformin reduces ATP production, which triggers cellular energy sensing pathways — most notably AMPK (AMP-activated protein kinase) activation. AMPK activation does produce some metabolically beneficial downstream effects, particularly in diabetic or insulin-resistant patients. This is the biochemical basis of the longevity claim.
However, here is the core problem: inhibiting Complex I directly impairs the mitochondrial electron transport chain — the very system whose dysfunction is the second of the three primary aging mechanisms. Rather than repairing or enhancing mitochondrial function, Metformin chemically suppresses it. For a population whose primary biological challenge is declining mitochondrial ATP output, deliberately reducing mitochondrial efficiency is counterproductive at best and actively harmful at worst.
Furthermore, Metformin is well-documented to cause Vitamin B12 depletion over time, a nutrient critical to DNA repair, neurological function, and red blood cell production. Long-term Metformin use without B12 supplementation compounds cellular dysfunction — directly worsening the genomic instability mechanism. Metformin also does not address cellular senescence or SASP in any meaningful, direct way. It touches one downstream pathway of one mechanism while actively impairing the infrastructure of another.
MOTS-C: The Mitochondria-Derived Peptide Targeting All Three Aging Mechanisms
MOTS-C (Mitochondrial Open Reading Frame of the 12S rRNA Type-C) is a mitochondria-derived peptide (MDP) — a 16-amino-acid peptide encoded not by nuclear DNA but by the mitochondrial genome itself, specifically within the 12S ribosomal RNA gene. It was first identified and characterized in 2015 and represents a fundamentally different category of biological signaling molecule compared to synthetic peptides or repurposed pharmaceuticals like Metformin.
What makes MOTS-C exceptional from a longevity research perspective is that it does not merely address one aging mechanism. Current research indicates it engages all three core mechanisms described above:
- Genomic Stability: MOTS-C activates AMPK and regulates the folate cycle and methionine metabolism, reducing the production of reactive oxygen species that drive DNA damage accumulation. By reducing mitochondrial ROS generation at the source, it addresses the primary driver of genomic instability.
- Mitochondrial Function: Unlike Metformin, which inhibits Complex I, MOTS-C enhances mitochondrial biogenesis and improves electron transport chain efficiency. It supports the production of new, functional mitochondria and promotes mitophagy — the cellular process that clears damaged mitochondria and replaces them with healthier ones.
- Cellular Senescence: MOTS-C has demonstrated senolytic and anti-inflammatory properties in research models, reducing SASP factor production and modulating the inflammatory environment that senescent cells create. By reducing the upstream ROS burden that triggers senescence checkpoints, it also reduces the rate at which cells are pushed into senescent states in the first place.
MOTS-C levels in humans decline naturally and significantly with age, which may partly explain why mitochondrial dysfunction and its downstream consequences accelerate after middle age. Exogenous MOTS-C administration in research models has demonstrated improvements in metabolic health, insulin sensitivity, physical endurance, cognitive function, and markers of systemic inflammation — without the mitochondrial suppression that characterizes Metformin's mechanism.
MOTS-C Dosing Protocol
The following table summarizes dosing information as discussed in current research contexts. Note: The source transcript for this article did not include specific dosing figures for MOTS-C or Metformin. The values below reflect commonly referenced research-based ranges from the broader MOTS-C literature and should be treated as general educational reference points only, not clinical prescriptions.
| Compound | Dose | Frequency | Timing | Route | Cycle |
|---|---|---|---|---|---|
| MOTS-C | Not specified in transcript | Not specified in transcript | Not specified in transcript | Not specified in transcript | Not specified in transcript |
| Metformin | Not specified in transcript | Not specified in transcript | Not specified in transcript | Oral (general knowledge) | Not specified in transcript |
| Vitamin B12 | Not specified in transcript | Not specified in transcript | Not specified in transcript | Not specified in transcript | Not specified in transcript |
Important note on MOTS-C dosing: The source transcript used to produce this article was truncated before specific dosing information was presented. Based on the broader published research and clinical research community consensus, MOTS-C is most commonly studied and used in research settings via subcutaneous injection, with doses typically ranging from 5 mg to 10 mg per injection, administered once to twice weekly. Some researchers explore daily microdosing protocols. Timing is frequently noted in the context of pre-exercise or morning administration to align with natural circadian mitochondrial activity patterns. Any individual considering
Shop Featured Peptides
Explore high-quality research peptides. Use our referral link to support independent research.
